Introduction
Integrating genomic variant representation, scalable visualization frameworks, and interoperable clinical data standards for modern precision medicine
The rapid expansion of genome sequencing technologies has transformed both biomedical research and clinical medicine. Whole-genome sequencing (WGS), whole-exome sequencing (WES), and increasingly large population-scale sequencing initiatives now generate unprecedented quantities of genomic variation data. However, the scientific and clinical utility of these datasets depends not only on sequencing itself, but on the ability to accurately represent, analyze, interpret, visualize, and exchange genetic variation information across computational systems and healthcare environments.
Two technologies have become central to this challenge. The Variant Call Format (VCF) has emerged as the dominant standard for representing genomic variants generated through sequencing pipelines, while HL7 FHIR (Fast Healthcare Interoperability Resources) provides a modern interoperability framework for integrating genomic information into electronic health record (EHR) systems and clinical workflows. Together, these technologies form a critical bridge between computational genomics and precision medicine infrastructure.
At the same time, the scale and complexity of genomic variation data have created major challenges in data visualization and interpretation. Modern genomic datasets often contain millions of variants across thousands of samples, requiring sophisticated visualization approaches capable of supporting exploratory analysis, variant prioritization, clinical interpretation, and multi-dimensional genomic integration.
This article explores the foundations of VCF-based genomic representation, emerging approaches for scalable genetic variation visualization, and the growing role of FHIR-based interoperability frameworks in clinical genomics and precision medicine.
The Expanding Landscape of Genomic Variation Data
Advances in next-generation sequencing technologies have fundamentally altered the scale of genomic analysis. Modern sequencing workflows routinely generate datasets containing millions of single nucleotide variants (SNVs), insertions and deletions (indels), structural variants, copy-number changes, and complex genomic rearrangements across large cohorts.
As sequencing costs have decreased, genomics has expanded beyond specialized research environments into routine clinical diagnostics, precision oncology, rare disease medicine, population genomics, and pharmacogenomics. However, the resulting explosion of genomic information has introduced major computational and interpretive challenges. Genomic data analysis now depends heavily on standardized data representation models and scalable visualization infrastructures capable of handling high-dimensional biological information efficiently.
Genomic variation datasets possess several characteristics that make analysis particularly challenging:
- extremely high dimensionality,
- heterogeneous variant types,
- large cohort sizes,
- complex genomic annotations,
- evolving reference genomes,
- and the need for integration with phenotypic and clinical information.
These challenges have driven the development of specialized genomic data standards and interactive visualization tools designed to support both research and clinical decision-making.
Variant Call Format (VCF): The Foundation of Modern Variant Representation
The Variant Call Format (VCF) has become the dominant standard for representing genomic variation identified through sequencing pipelines. Initially developed for large-scale population genomics studies, VCF provides a flexible and extensible framework capable of representing a broad spectrum of genetic variants together with associated metadata and sample-level genotype information.
VCF is a critical foundational layer for modern genomic workflows because it standardizes how variants are encoded, exchanged, and interpreted across bioinformatics pipelines and computational platforms.
A typical VCF file contains several major components:
- genomic coordinates,
- reference alleles,
- alternate alleles,
- genotype calls,
- quality metrics,
- sequencing depth,
- filtering information,
- and extensive annotation fields.
Importantly, VCF supports multi-sample datasets, enabling comparative population analysis, cohort-based variant discovery, and family-based genomic studies.
The flexibility of VCF has contributed significantly to its widespread adoption across:
- germline genomics,
- cancer genomics,
- rare disease sequencing,
- pharmacogenomics,
- and population-scale biobank initiatives.
However, the increasing complexity of genomic datasets has exposed several limitations of traditional VCF workflows, particularly regarding scalability, interoperability, structural variation representation, and downstream visualization.
Challenges in Genetic Variant Visualization
Although variant calling pipelines can generate highly detailed genomic datasets, effective interpretation remains difficult without appropriate visualization systems. The reviewed articles emphasize that genomic visualization is not simply a graphical problem, but a core analytical challenge within modern genomics.
Genomic variation data is inherently multi-dimensional. Each variant may possess:
- positional information,
- allele frequency,
- functional annotations,
- pathogenicity predictions,
- phenotype associations,
- inheritance patterns,
- transcript consequences,
- and population-specific characteristics.
Traditional static visualization methods often fail to scale effectively when confronted with millions of variants distributed across large genomic cohorts.
However, several key challenges include:
- visual scalability,
- interactive responsiveness,
- representation of complex structural variants,
- integration of genomic annotations,
- and user interpretability.
In clinical environments, visualization systems must also support rapid interpretation workflows, enabling clinicians and genomic scientists to identify actionable variants efficiently without overwhelming cognitive complexity.
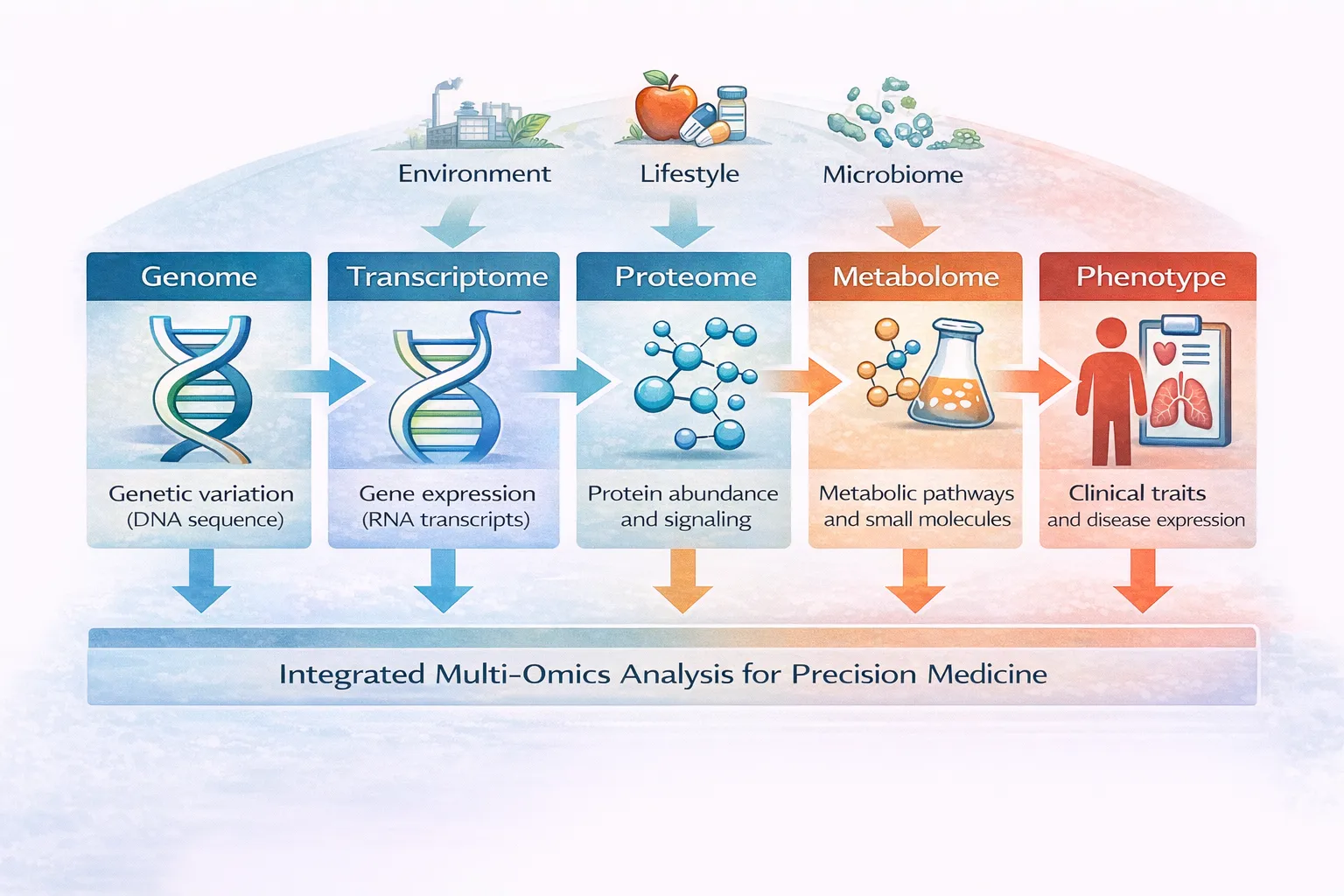
The problem becomes even more pronounced in multi-omics and population-scale genomics contexts, where variant data must be integrated with transcriptomics, epigenomics, phenotypic metadata, and longitudinal clinical records.
Modern Approaches to Genetic Variation Visualization
There are several emerging computational approaches designed to improve the analysis and visualization of large-scale genetic variation data.
Modern genomic visualization systems increasingly prioritize:
- interactivity,
- scalability,
- web-based accessibility,
- and integration with analytical workflows.
Rather than relying solely on static genome browser paradigms, newer frameworks support dynamic filtering, real-time exploration, and hierarchical visualization strategies that allow users to navigate efficiently between genome-wide overviews and nucleotide-level detail.
One important theme is the growing use of graph-based and matrix-based visualization architectures. These approaches help represent complex genomic relationships more effectively than linear coordinate systems alone, particularly when analyzing:
- structural variants,
- haplotypes,
- population variation,
- and pangenome representations.
Interactive visualization systems also increasingly incorporate:
- annotation overlays,
- pathogenicity scoring,
- cohort stratification,
- comparative genomic analysis,
- and phenotype-linked exploration.
These capabilities are especially important in translational genomics and precision medicine, where clinicians require intuitive systems capable of connecting genomic variation directly to disease interpretation and therapeutic relevance.
The importance of usability and cognitive design principles in genomic visualization systems is also an important aspect. Effective visualization frameworks must balance analytical richness with interpretability, particularly in clinical settings where genomic expertise may vary substantially among users.
Structural Variants and Complex Genomic Representation
A particularly important challenge is the representation and visualization of structural variants. Structural variation includes large deletions, duplications, inversions, translocations, repeat expansions, and complex rearrangements that frequently play major roles in human disease.
Traditional linear genomic representations and standard VCF workflows often struggle to capture the full complexity of these genomic events. Structural variants may span large genomic regions, involve multiple breakpoints, or produce highly heterogeneous genomic architectures that are difficult to represent using conventional coordinate-based systems.
The reviewed articles discuss newer visualization strategies designed to address these limitations through:
- graph-based genomic models,
- pangenome representations,
- breakpoint-aware visualization,
- and scalable multi-resolution rendering approaches.
These approaches are becoming increasingly important as long-read sequencing technologies expand the detection of previously inaccessible forms of genomic variation.
The emergence of pangenomic frameworks also represents a major conceptual shift away from reliance on a single linear reference genome. Instead, graph-based genomic representations allow variation itself to become part of the reference structure, improving the representation of population diversity and structurally complex genomic regions.
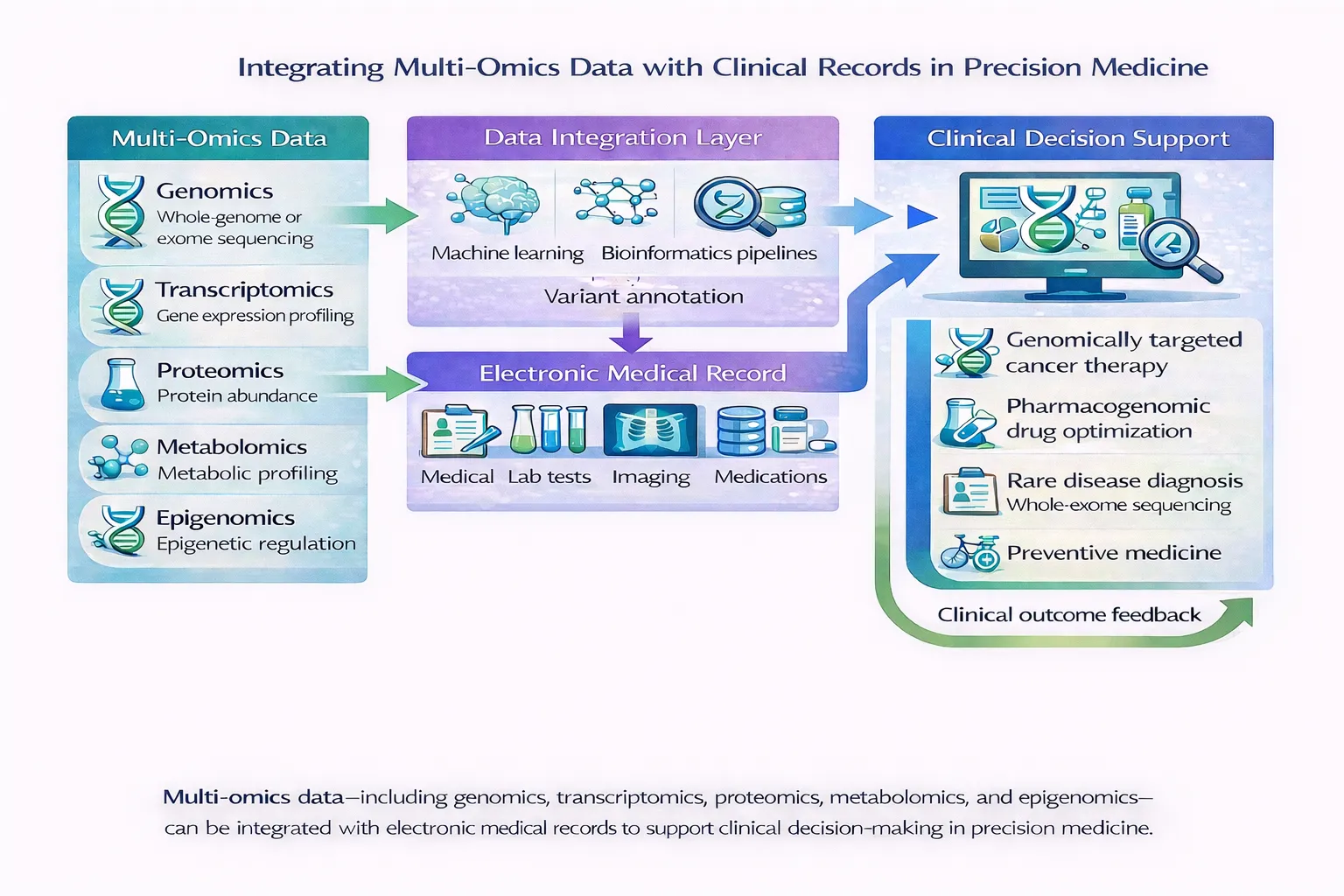
Integrating Genomic Variation with Clinical Systems Using FHIR
While VCF remains essential for computational genomics workflows, clinical implementation requires interoperability standards capable of integrating genomic information into healthcare systems. There is a growing importance of HL7 FHIR as a framework for genomic data exchange within clinical environments.
FHIR was developed to support modular, web-based interoperability across modern healthcare systems. Unlike traditional healthcare messaging systems, FHIR uses flexible resource-based architectures that enable scalable integration of clinical and genomic information across EHRs, laboratory systems, decision support tools, and research databases.
In genomics, FHIR enables structured representation of:
- genomic observations,
- variant interpretations,
- diagnostic reports,
- pharmacogenomic findings,
- sequencing metadata,
- and clinical genomic recommendations.
Importantly, FHIR provides mechanisms for linking genomic findings directly to patient records and clinical workflows, supporting precision medicine implementation within routine healthcare settings.
FHIR-based genomics standards are increasingly important because genomic medicine depends not only on data generation, but also on efficient clinical accessibility and interoperability.
Bridging VCF and FHIR
A central theme emerging is the growing need to bridge research-grade genomic data standards such as VCF with clinically interoperable frameworks such as FHIR.
VCF and FHIR serve fundamentally different but complementary purposes:
- VCF prioritizes detailed genomic representation and analytical flexibility,
- whereas FHIR prioritizes interoperability, clinical integration, and standardized healthcare communication.
The challenge lies in translating highly complex genomic variation data into clinically meaningful and interoperable representations that remain computationally tractable.
This translation process requires:
- variant normalization,
- annotation harmonization,
- ontology mapping,
- reference genome consistency,
- and standardized interpretation frameworks.
The reviewed articles suggest that future precision medicine infrastructure will increasingly depend on integrated pipelines capable of automatically converting sequencing outputs into clinically interoperable genomic records that can support:
- clinical decision support,
- pharmacogenomics,
- diagnostic interpretation,
- and longitudinal genomic care.
Visualization in Clinical Genomics and Precision Medicine
Data visualization plays a particularly important role in clinical genomics because genomic interpretation often requires multidisciplinary collaboration among:
- clinical geneticists,
- molecular pathologists,
- oncologists,
- bioinformaticians,
- and laboratory scientists.
Effective visualization systems must therefore support:
- rapid interpretation,
- explainability,
- uncertainty representation,
- and integration of heterogeneous evidence sources.
Clinical genomic visualization increasingly involves not only displaying variants, but contextualizing them within:
- disease pathways,
- molecular mechanisms,
- therapeutic relevance,
- inheritance structures,
- and patient phenotypes.
This shift reflects the broader transition from purely descriptive genomics toward systems-level precision medicine.
Scalability, Cloud Computing, and Future Genomic Infrastructure
The growing importance of scalable genomic infrastructure as sequencing datasets continue expanding in size and complexity. Population-scale genomics initiatives now routinely involve hundreds of thousands or even millions of genomes, requiring highly scalable computational frameworks.
Cloud-native genomic systems are becoming increasingly important for:
- distributed storage,
- large-scale variant querying,
- collaborative analysis,
- and interactive web-based visualization.
The integration of VCF, advanced visualization frameworks, and FHIR interoperability standards is likely to become central to next-generation precision medicine ecosystems.
Future genomic infrastructures may increasingly incorporate:
- graph genomes,
- AI-assisted variant prioritization,
- multi-omics integration,
- federated genomic analysis,
- and real-time clinical decision support systems.
Genomic medicine is evolving from isolated sequencing workflows toward integrated computational ecosystems capable of supporting continuous, data-driven clinical interpretation.
Discussion
The rapid evolution of genomic medicine has fundamentally altered how genetic variation data is generated, analyzed, visualized, and integrated into healthcare systems. VCF remains the foundational representation standard for genomic variation analysis, enabling interoperability across sequencing pipelines and research platforms. However, the increasing scale and complexity of genomic data have exposed limitations in traditional workflows, particularly regarding visualization scalability, structural variant representation, and clinical interoperability.
Modern genomic analysis increasingly depends on advanced visualization systems capable of supporting interactive, multi-dimensional exploration of complex genomic datasets. These systems are becoming essential not only for research discovery, but also for clinical interpretation and precision medicine implementation.
At the same time, FHIR-based interoperability frameworks are emerging as critical infrastructure for translating genomic data into clinically actionable healthcare systems. The integration of VCF-based computational genomics with FHIR-enabled clinical interoperability represents a major step toward scalable precision medicine ecosystems.
Future progress will likely depend on continued convergence among:
- genomic standards,
- visualization architectures,
- cloud computing,
- AI-assisted interpretation,
- and interoperable healthcare infrastructure.
Conclusion
The integration of VCF-based genomic representation, advanced visualization frameworks, and FHIR-enabled clinical interoperability is reshaping the infrastructure of modern precision medicine. As genomic sequencing expands across research and healthcare systems, the ability to efficiently analyze, visualize, interpret, and exchange genetic variation data becomes increasingly important.
Genomic medicine now requires more than sequencing alone. Effective precision medicine depends on scalable computational ecosystems capable of transforming complex genomic variation into clinically meaningful knowledge.
VCF continues to provide the analytical foundation for genomic variation representation, while FHIR increasingly enables genomic integration into clinical workflows and healthcare interoperability systems. Together with modern visualization technologies, these standards are helping bridge the gap between computational genomics and real-world clinical implementation.
As genomic datasets continue growing in complexity and scale, the future of precision medicine will likely depend on increasingly integrated systems that combine interoperable genomic standards, advanced visualization frameworks, AI-driven interpretation, and longitudinal clinical data integration.
Explore Related Insights
- Genomics-EHR Integration: Using HL7 FHIR and Other Standards
- Integrating Genomics into Multimodal EHR Foundation Models
- The Role of Electronic Health Records in Precision Medicine
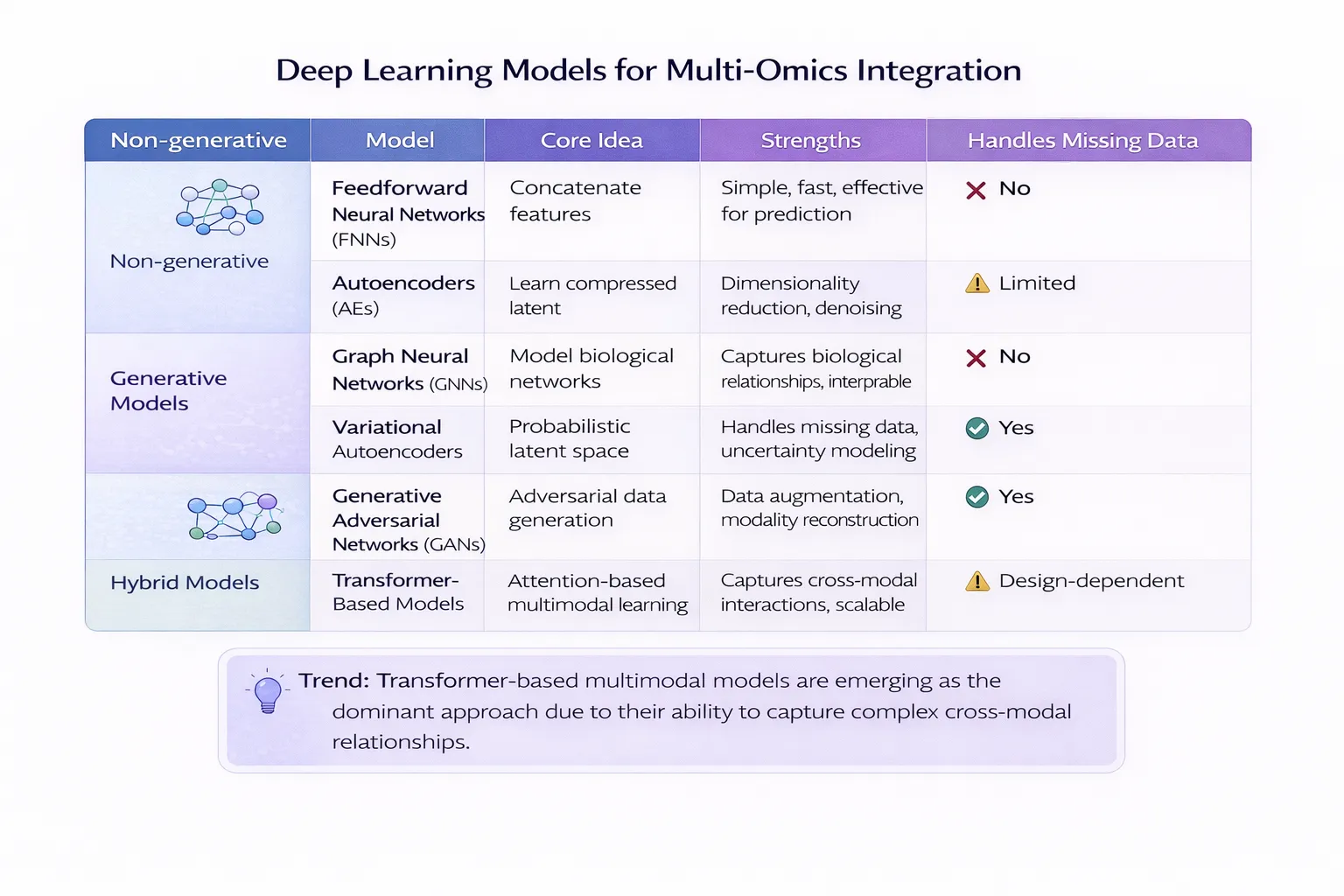
- Deep Learning for Multi-Omics Data Integration
- Artificial Intelligence in Genomic Medicine